Ангиокератомы. Виды ангиокератом. Диагностика и лечение ангиокератом.
Ангиокератомы являются капиллярными сосудистыми мальформациями. Они образованы стойкими субэпидермальными капиллярными расширениями) и изменениями эпидермиса с явлениями папиллома-тоза, гиперкератоза и акантоза.
Различают несколько клинических форм ангиокератомы: ограниченную ангиокератому, невоидную ангиокератому пальцев Мибелли, солитарную папулезную ангиокератому мошонки (вульвы) Фордайса, диффузную ангиокератому туловища Фабри. Только ограниченная ангиокератома присутствует с рождения, У некоторых больных могут наблюдаться элементы более чем одного типа. Очаги ангиокератомы иногда являются признаком синдромов Клиппеля—Тренонея—Вебера и Кобба.
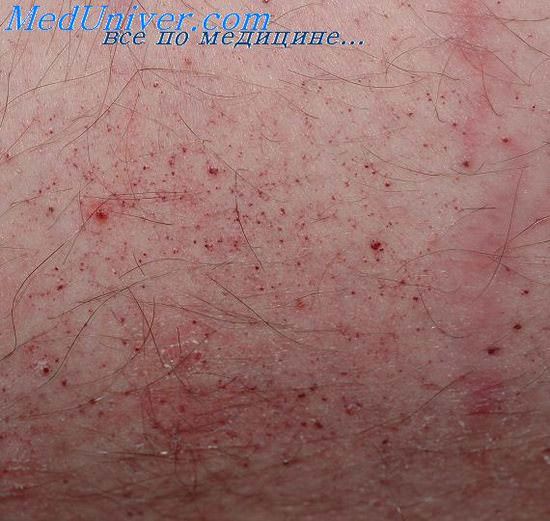
Обычно ангиокератомы возникают в виде мягких узелков розового или красного цвета, диаметром от 1 до 3 мм. Со временем во многих из них развиваются кератотические изменения, они становятся темнее и приобретают сходство с бородавками.
Ангиокератома ограниченная представляет собой гиперкератотический сосудистый узелок диаметром 2-5 мм, локализующийся на коже бедра, голени или стопы. Цвет узелка варьирует от темно-красного до сине-черного. Конфигурация неправильная. При легкой травме кровоточит. Гистологическое строение типичное.
Ангиокератома ограниченная невоидная пальцев Мобелли (син.: ангиокератома Мибелли) — наследственная форма ангиокератом с аутосомно-доминантным типом наследования. Болеют чаше девочки, страдающие акроцианозом, ладонно-подошвенным гипергидрозом. Первое проявление заболевания возникает обычно в возрасте 10-15 лет и характеризуется мелкими (точечными) сосудистыми красными пятнами на коже пальцев кистей и стоп. Высыпания медленно увеличиваются (до 5 мм в диаметре), приподнимаясь над уровнем кожи, цвет их становится более темным, на поверхности появляются роговые наслоения. Иногда ангиокератома Мибелли осложняется изъязвлением.
Ангиокератома солитарная папулезная обычно развивается в возрасте от 10 до 40 лет вследствие травматизации ограниченной ангиокератомы. При этом последняя резко увеличивается в размере до 10 мм, приобретая вид узелка с бородавчатой поверхностью, возвышающегося над окружающей кожей, цвет ее темно-красный или сине-черный. Сине-черные элементы следует дифференцировать с меланомой.
Ангиокератома мошонки (вульвы) Фордайса (ангиокератома Фордайса) появляется в более позднем возрасте (от 16 до 70 лет). Ее развитию способствуют локальный венозный стаз, дистрофические изменения эластических волокон кожи мошонки. Клинически характеризуется мелкими (1-4 мм в диаметре) ярко-красными сосудистыми узелками, которые постепенно увеличиваются в размере, и окраска их становится более темной. Высыпания часто расположены вдоль поверхностных вен. Ангиокератомы мошонки могут ассоциироваться с варикоцеле, паховой грыжей или тромбофлебитом.
Ангиокератома вульвы встречается значительно реже, чем ангиокератома мошонки, и локализуется на больших половых губах, глав ным образом у пожилых женщин. У молодых женщин ангиокератомы вульвы могут развиваться в связи с повышением венозного давления во время беременности или использованием оральных контрацептивов.
Ангиокератома Фабри (син.: Фабри диффузная ангиокератома туловища, липоидоз дистопический наследственный, гликосфинголипидоз. перамидтригексозидоз) — наследственная ферментопати], проявляющаяся развитием множественных ангиокератом кожи и слизистых оболочек с поражением глаз и внутренних органов (почки, сердце и др.). В основе развития болезни лежит нарушение обмена гликолипидов в результате снижения активности или полного отсутствия фермента церамидтригексозидазы (альфгалактозидазы), что ведет к накоплению в тканях липида церамидтригексозида. Тип наследования рецессивный, сцепленный с Х-хромосомой. Заболевание проявляется у мальчиков в юношеском возрасте и постепенно прогрессирует, приводя к почечной или сердечной недостаточности. Клинически характеризуется возникновением на коже и слизистых оболочках множественных кератинизированных сосудистых узелков величиной с булавочную головку темно-красного цвета, локализующихся на губах, щеках, в подмышечных ямках, пупочной области, на мошонке, пальцах.
Гистологически выявляют расширение капилляров, часто они имеют вид внутридермальных «кровяных кист». Гистохимически в коже, как и в других органах, обнаруживают отложения фосфолипидов и церамидтригексозида. Наблюдаются ампулоподобные расширения коньъюнктивальных вен, участки помутнения роговицы в виде веерообразных полос в радужке — узелки спонтанной гифемы, особенно у детей грудного возраста, характерны также гетерохромия радужки, глаукома.
При осмотре глазного дна выявляют симптом «медной проволоки», обусловленный расширением и извитостью вен сетчатки, гиперемию дисков зрительных нервов, микроаневризмы сосудов сетчатки. Тригексоцерамид откладывается в гладких мышцах и эндотелии сосудов радужки, цилиарного тела, хориоидеи и орбиты, в эпителиальных клетках конъюнктивы, капсуле хрусталика и других тканях глаза. В процесс вовлекаются внутренние органы: обнаруживается вакуолизация клеток клубочков и канальцев (вплоть до развития уремии) почек, миокардит и т.д. В 20% случаев возникают симптомы полиневрита, больных беспокоят боли в конечностях, животе (вплоть до картины «острого живота»). В крови и моче выявляют накопления тригексоцерамида.
Диффузная ангиокератома туловища не является исключительно проявлением болезни Фабри; она также описана в связи с другими редкими наследственными болезнями накопления — например с фукозидозом II типа, галактозидозом II типа, болезнью Канзаки, или дефицитом альфа-1М-ацетилга-лактозаминидазы, спартилгликозаминурией, сиалидозом II типа. С другой стороны, диффузная ангиокератома туловища описана и при отсутствии метаболических нарушений.
При ангиокератоме Фабри важное диагностическое значение имеет выявление вакуолизации эндотелиальных и гладкомышечных клеток в артериолах и мышцах, поднимающих волос. Однако ее не всегда удается обнаружить в срезах, окрашенных гемоатоксилином и эозином, и легче обнаружить при окраске препаратов Суданом черным В или при ШИК-реакции. При этом следует учитывать, что отложения гликолипидов в вакуолях могут быть не только у больных диффузной ангиокератомой туловища, но и у больных, страдающих сходными заболеваниями. Однако отложение гликолипидов у больных ангиокератомой Фабри имеются не только в ангиокератомах, но и в видимо здоровой коже. Кроме того, при ангокератоме Фабри при электронной микроскопии крупные отложения жира обнаруживают в эндотелиальных клетках, перицитах, фибробластах, мышцах, поднимающих волос, а также в секреторных проточных и миоэпителиальных клетках эккринных желез. Эти отложения могут иметь характерную пластинчатую структуру, и их не видно при других разновидностях ангиокератом или в элементах ангиокератомы Фабри, протекающей без ферментных нарушений.
Диагноз ангиокератомы основывается на клинических данных, подкрепленных в сомнительных случаях результатами гистологического исследования, а ангиокератомы Фабри — также на обнаружении скоплений церамидтригексозида в крови, биоптатах почек и моче.
Диффернциальный диагноз ангиокератомы проводится с веррукозной гемангиомой, для которой характерна ангиоматозная (капиллярная) эндотелиальная пролиферация, лимфангиомой и меланомой; при ангиокератоме Фабри — с болезнью Рандю—Ослера.
Прогноз ангиокератомы неблагоприятный, у гомозиготных мужчин возможен летальный исход к 40 годам от уремии или инсульта.
Лечение ангиокреатом хирургическое; проводятся также удаление лучами лазера, элекрокоагуляция, криотерапия отдельных элементов.